
The following information presented is based on the author’s interpretation and understanding of the EudraLex Volume 4 Annex 15 – Qualification and Validation, specifically on the topic of qualification stages for equipment. The scope of the blog post is on Installation Qualification (IQ), Operational Qualification (OQ) and Performance Qualification (PQ). The definition of each of the qualification stages will be illustrated by using a -20°C freezer that is used to store GMP raw materials.
For discussion, let’s assume that a risk-based Commissioning, Qualification and Validation (CQV) approach following the guidance of ASTM 22500–13 was used to demonstrate that the -20°C freezer is fit for intended use, i.e. maintaining an environment condition suitable for storing temperature-sensitive GMP raw materials.
The following diagram summarises the process as described in the ASTM E2500–13 “Standard Guide for Specification, Design, and Verification of Pharmaceutical and Biopharmaceutical Manufacturing Systems and Equipment.”
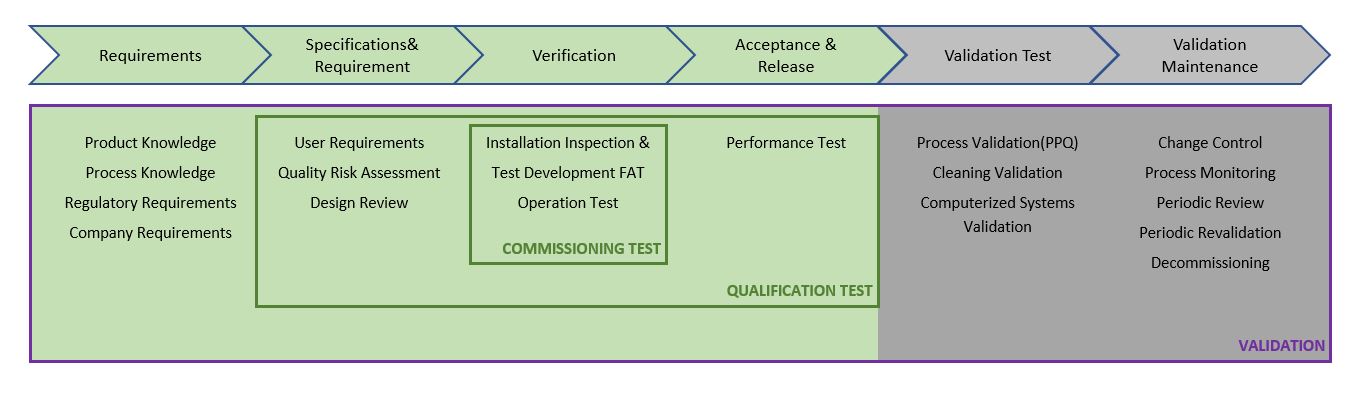
Figure 1: ASTM E2500-13 Process for Specification, Design and Verification
Note that the ASTM E2500–13 uses the term “verification” instead of qualification. The term “verification” is defined as a systematic approach to verify that manufacturing systems, acting singly or in combination, are fit for intended use, have been properly installed, and are operating correctly. This is an umbrella term that encompasses all types of approaches used to ensure that systems are fit for use such as qualification, commissioning and qualification, verification, system validation, or others.
Installation Qualification (IQ)
In generic terms, Installation qualification (IQ) can be defined as a documented verification process that the system (facilities, utilities, equipment – FUE) has been properly delivered, installed and configured according to a pre-approved set of acceptance criteria. Using the -20°C freezer as an example, the IQ requirements cover, amongst others, the following:
- The delivered and installed -20°C freezer model is per the equipment datasheet.
- The -20°C freezer is tagged per the equipment datasheet.
- The -20°C freezer is installed at its Intended installation location.
- The -20°C freezer is installed following the manufacturer-recommended installation requirements, for e.g. environmental requirements that the -20°C freezer could operate optimally without damaging the compressor, distance from the back wall, and the adjacent equipment for ease of maintenance and heat dissipation, etc.
- The -20°C freezer is connected to the appropriate power source, for e.g. the power supply is 230V/1Ph/50Hz with emergency power supply, etc.
- The -20°C freezer temperature control element(s) is calibrated.
- Availability of required documents, i.e. instruction manual, drawings, etc.
- Etc.

Operational Qualification (OQ)
Depending on the CQV strategy developed and documented in the CQ Execution Plan (CQEP), the Operational Qualification (OQ) could be started together with the IQ, with a prerequisite of specific IQ test items performed OR could be more conservative, requiring that the IQ document is post-approval prior to the start of OQ. In generic terms, OQ can be defined as documented testing of the system (facilities, utilities, equipment – FUE) per specification as listed by the manufacturer of the equipment and/or design specification.
Depending on the CQ approach that is adopted by the project, certain OR all aspects of the system will be tested against pre-approved acceptance criteria and documented. Using the -20°C freezer as an example, the OQ requirements cover, amongst others, the following:
- Noise during operation does not exceed local authority limits.
- Functionalities verification that all critical operation features are operating as intended. The functionalities could include, but not limited to, buzzer, indication lights, critical alarms, blackout response, etc.
- Empty Chamber Temperature Mapping to ensure that the -20°C freezer could operate within the intended temperature range(s).
- Etc.
One should be clear on the scope of the OQ and its objective of verifying that the system (facilities, utilities, equipment – FUE) meets the specification as listed by the manufacturer of the equipment, and/or design specification. Challenges to the system are not normally part of the OQ.
At the conclusion of the OQ, the -20°C freezer is deemed qualified for meeting all the applicable requirements in the User Requirement Specification, and could be released to the next phase of the process, which is Performance Qualification (PQ). Any changes arising post-approval of OQ is normally managed through the organization Change Control SOP, which mandates Quality Assurance involvement.
Performance Qualification (PQ)
PQ should normally follow the successful completion of IQ and OQ. However, it may in some cases be appropriate to perform it in conjunction with OQ depending on the CQ strategy of the project.
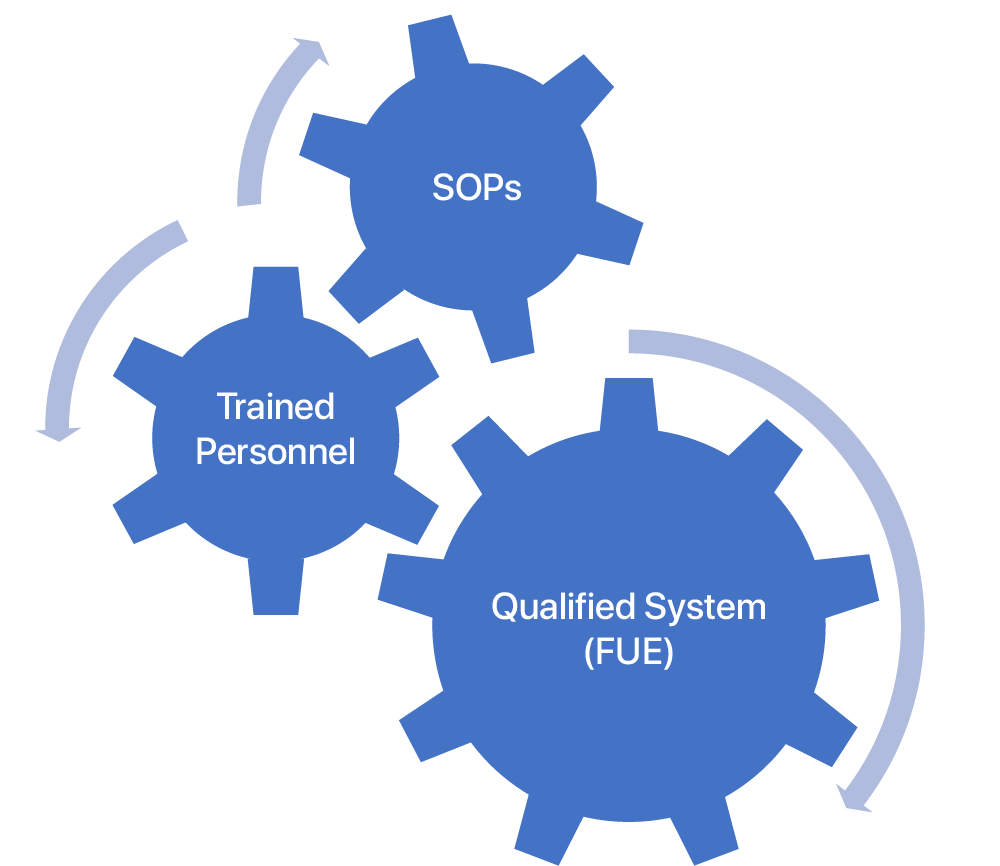
The PQ process integrates procedures, personnel, and the qualified (IQ/OQ/IOQ-ed) systems and is put through specifically designed verification tests to ensure that the system consistently and repeatedly meets the expected results.
Using the -20°C freezer as an example, the PQ tests may include, but not limited to the following studies:
- Loaded chamber temperature mapping
- Power outage study
- Open door study
- Temperature recovery study
- Etc.
The PQ studies cited above identify the variables (duration, material, the quantity of material, etc.) and aim to identify the range(s) within which these variables should fall, to ensure that the -20°C freezer consistently and repeatedly performs such that the GMP raw materials are stored under intended temperature without excursion. All the setpoints gathered from the PQ studies shall be the basis of routine GMP operations. Any changes to them shall be governed by the organisation’s change control procedure.
You can read this article if you’re interested in becoming a CQV Engineer in the pharma industry, which offers a few points for your consideration.
At No deviation, we partner with you to help you with executing your CQV process, troubleshooting or your CAPA, or implementing new digital solutions. Our team will come with an open mind and follow the simple steps that are Understand, Observe, Define, and Implement.
Reach out to us for a full risk-based paperless validation with integrated commissioning and test plan or to bring efficiency to your existing paper-based IQ, OQ execution. You can also visit our website for more information on our services.