

How many of us have seen this circulating comic strip and smiled to ourselves, knowing that this is exactly the situation at work – whether you are the engineer or the user? I am using this example as an opening to the topic as most of us can relate to it at the most basic level. In this blog post, I will try to explain the what, when, where, who, why and how of Design Qualification.
The following information presented is based on the author’s interpretation and understanding of the EudraLex Volume 4 Annex 15 – Qualification and Validation, specifically on the topic of qualification stages for equipment.
For continuity of discussion from our post on IQ, OQ, and PQ in the pharmaceutical industry, let us assume that a risk-based Commissioning, Qualification and Validation (CQV) approach following the guidance of ASTM 2500–13 was used to demonstrate that the -20°C freezer is fit for intended use, i.e. maintaining an environment condition suitable for storing temperature-sensitive GMP raw materials.
1. Why is Design Qualification important?
DQ provides documented verification that the design of a new or modified direct impact system will result in a system that is suitable for the intended purpose. The sole and most important objective of DQ is to:
- Verify that the CAs/CDEs necessary to adequately control risks to product quality and patient safety as identified during the Quality Risk Assessment (QRA) are present in the design.
The practice of DQ is essentially a quality assurance process to ensure that the equipment will meet its intended use. It would be costly, from a time and monetary perspective, if a piece of equipment is found to not have the required CAs/CDEs to mitigate the risks identified during commissioning – or worse, during qualification state.
2. What is Design Qualification (DQ) and how does it relate to Design Review (DR)?
The introductory paragraph to Design Review and Design Qualification in Section 5 of the ISPE Baseline Guide Volume 5: Commissioning and Qualification Second Edition, summarizes concisely the relationship between Design Review and Design Qualification as follows:
“DR and DQ are not intended to be separate activities, but rather separate documentation in which DQ is focused on Critical Aspects (CAs)/Critical Design Elements (CDEs) and involves the Quality Unit as an approver. There should be minimal duplication of work. The final report from DR is a key input into the DQ process.”
Having squared away the relationship between DR and DQ, what exactly then is DQ?
What is meant in the above paragraph that DQ is focused on the Critical Aspects (CAs)/Critical Design Elements (CDEs)? To answer that, let us look at the relational diagram for C&Q activities based on the ISPE Baseline Guide Volume 5: Commissioning and Qualification Second Edition as follows:
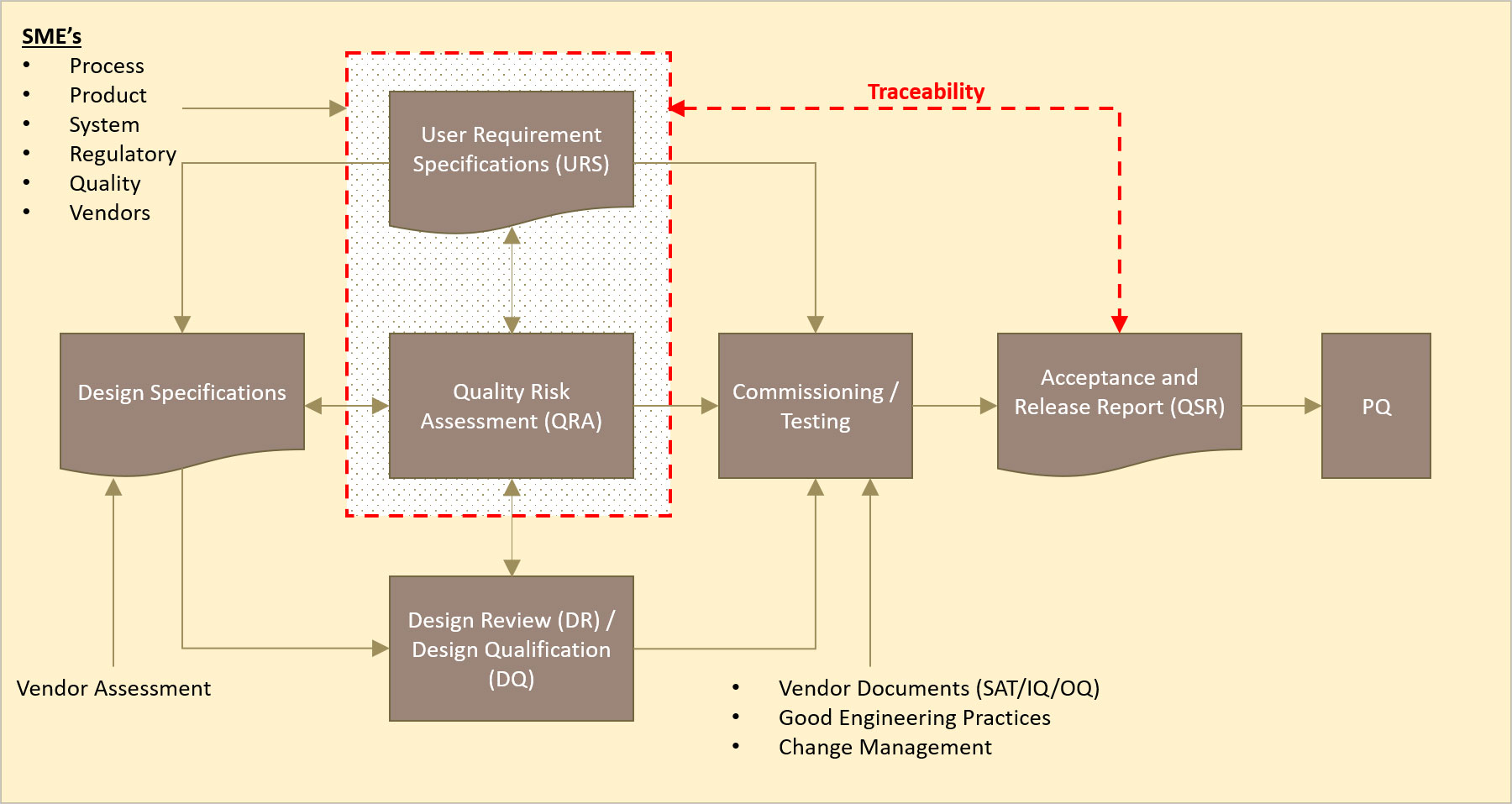
QRA is applied to a direct impact system to examine the product quality risk controls. This assessment identifies the critical design controls (CAs/CDEs) and procedural controls that are required to mitigate system risks to an acceptable level. The critical design controls (CAs/CDEs) are documented in the URS.
Simplistically, the DR is an engineering deliverable that ensures that all aspects of the URS are checked against the design specification from the various vendor submissions – including quality requirements, business requirements, Environmental, Health and Safety (EHS) requirements and others.
The DQ is then a “carved out” documentation that focuses on the quality requirements i.e. the critical design controls (CAs/CDEs), which typically require approval from the Quality Unit.
For off-the-shelf equipment, depending on the complexity and/or risk, regarding the patient and/or product quality, a DQ may not be necessary. This strategy however shall be clearly defined in the CQ Execution Plan through consultation with the Quality Unit and SMEs.
Taking the -20°C freezer, the Critical Quality Attribute (CQA) of the equipment is the ability of the equipment to maintain the environment condition at -20°C. In this case, the CA/CDE would simply be a freezer that could provide an environment at that -20°C with tolerance specified in the URS. There was no customization or whatsoever to the equipment. In that regard, for as long as the vendor-proposed equipment specification meets the CA/CDE, this URS point is met.
Therefore, there is no real value in performing a DQ. A simplified DR could quite easily meet the intent.
3. When should Design Qualification be executed?
It is important to reiterate that the DQ is not a separate activity from the DR but merely an outcome of the DR. The better question would be when DR should be executed.
The DR is performed generally in 3 stages:
(1) During the development of the concept
(2) Basis of Design (BoD) packages, and
(3) Near the completion of detailed design.
With every stage, there could be reiteration of the URS with developing information gathered from various sources such as process development, the introduction of better technology/solutions from vendor offerings etc. These design packages should be reviewed to ensure all URS (Business, Quality, and EHS) points are met.
Once the design review report or equivalent (depending on the complexity of the project) is completed, the DQ documentation could be started and concluded with the approval from the Quality Unit. This is an important point in time for a direct impact system as any changes to the equipment here forth will need to be managed through a systematic change evaluation process with the Quality Unit as a stakeholder.

4. Who should be taking part in the Design Qualification?
As DQ is an outcome of the DR, the presence and participation of the right stakeholders in the DR process will ensure a smooth and successful DQ.
As stated earlier, DR is an engineering deliverable and is a precursor to a successful DQ. DR examines to ensure all aspects of the URS (engineering design and quality aspects) are checked against the design specification.
As a minimum,
(1) Technical SMEs,
(2) System owners, and
(3) Any other stakeholders
having interests in the technical aspects of the system including operability, maintainability, and safety, should be present during the DR. Quality function is recommended but not mandatory to partake in the DR exercise to ensure smooth preparation of the DQ.
5. How is Design Qualification executed?
The most important precursory step is that the strategy for DQ is documented in the CQ Execution Plan and approved by the Quality Unit.
Assuming that the equipment has been assessed and needs to have a DQ performed, the prerequisites for DQ include:
- Identification of the correct reviewers/signatories for the DQ
- Approved URS that includes design (CAs/CDEs) controls that will mitigate the risks identified
- Approved DR(s)
Each CAs/CDEs point derived from the QRA that is in the approved URS will be mapped against objective evidence that the CAs/CDEs is/are present in the proposed design solution from the engineers and/or vendors. The documentation of the verification can be varied but the fundamental concept remains the same.
6. Where should the Design Qualification be recorded?
Depending on the (1) complexity of the project and (2) strategy adopted by the project, there are various ways by which the Design Qualification could be recorded. This strategy however shall be clearly defined in the CQ Execution Plan in consultation with the Quality Unit and the SMEs.
Based on experience in projects, it could be one of the following:
- As a standalone DQ document that makes cross-references to URS quality critical points.
- As an integrated section within the URS. The URS will be revised near the end of the detail design stage to include the references to design specifications/documentation demonstrating that the URS points (CAs/CDEs) are met.
- As an integrated section within the Traceability Matrix. The CAs/CDEs answering to each of the CPPs and subsequently the CQAs are mapped out in one revision of the Traceability Matrix. Upon completion of the verification process, the Traceability Matrix is once again revised to include the references to the verifications performed.
In whichever manner that the DQ is documented, it should contain the explicit statement that the design is suitable for the intended purpose and the report should be approved by representatives from applicable departments and the Quality Unit as a mandatory signatory.
At No deviation, we partner with you to help you with executing your CQV process, troubleshooting or your CAPA, or implementing new digital solutions. Reach out to us for a full risk-based paperless validation with integrated commissioning and test plan or to bring efficiency to your existing paper-based IQ, OQ execution.
You can also visit our website for more information on our services.
Pingback: Unit – II – “Being beyond the best”